研究テーマ
第一原理計算を用いた計算材料データベースの構築
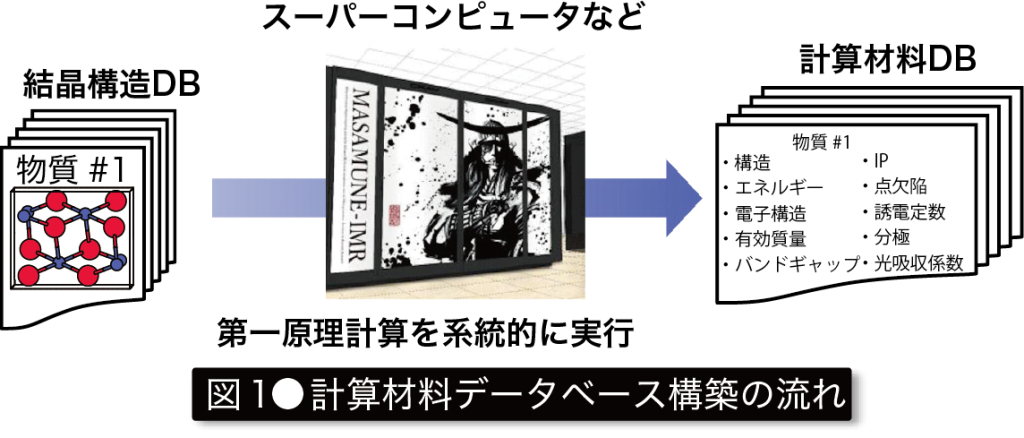
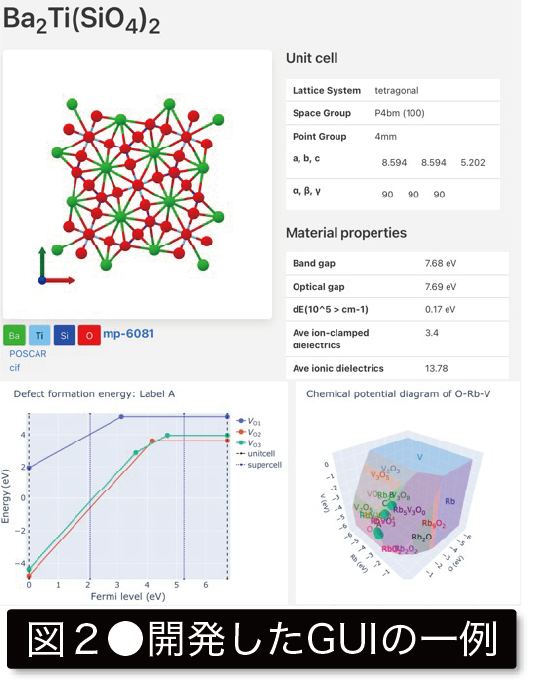
従来の材料研究は、実験主体で仮説検証を繰り返す中で、新材料の発見を行ってきました。しかし合成・計測を伴う実験過程は、多くの資金や人的資本を必要とし、開発速度に大幅な制約がかかります。一方、計算機はムーアの法則に従い、指数関数的な凄まじい成長により著しく進展し、その結果、量子力学に基づき物性を予測する第一原理計算を材料研究で用いることが可能となりました。さらに最近では、数十万物質を対象に第一原理計算を行い、得られた物性をデータベース(DB)化することが可能となりました(図1)。このように材料学においてもビッグデータの時代を迎えています。 この計算材料DBには、「既報物質の中から、用途に応じて最良の材料を選抜できる」、「機械学習(ML)の学習用データとして用いることができる」、「多数の物質から、物理・化学的に特異な傾向を示す物質を発見できる」など多数の活用法があります。本研究グループでは、計算材料DBに必要な基盤技術を構築し、それらを応用することで革新的新材料の発見と教科書に載るような物性の起源解明を目指しています(図2)。
物質中の点欠陥を対象とした第一原理計算
物質中には、空孔や不純物元素などの点欠陥が多数存在し、それらが材料機能発現において重要な役割を担っています。例えば、2014年に赤崎先生らがノーベル物理学賞を受賞したp型GaNは、僅かなMgを添加することにより実現されました。また、2019年に吉野先生らがノーベル化学賞を受賞したLiイオン伝導体の負極材料においては、Liの拡散のために多量のLi空孔を導入する必要があります。しかしながら、点欠陥特性を詳細に調べるために必要な原子レベルでの解析を、実験単独で行うことは極めて難しいです。そこで2000年頃より、第一原理計算を用いて点欠陥特性を調べる研究が行われる様になりました。
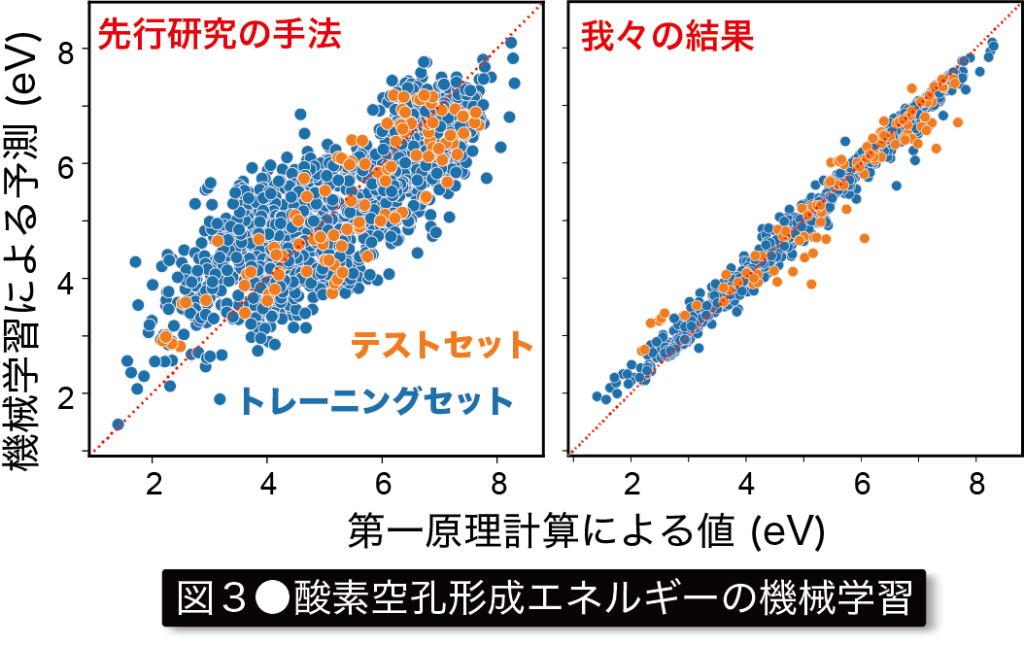
私達はこれまで、点欠陥の形成エネルギーや吸収発光に伴う遷移エネルギーなどを高精度に計算する手法の開発や、複雑な点欠陥計算の処理を自動化するためのプログラム開発を行い、それらを様々な物質へと応用してきました。また最近では、上述した計算材料データベース開発のプログラムを併用することで、およそ1,000酸化物中の酸素空孔を対象とした系統的計算を行い、その空孔形成エネルギーを対象とした機械学習などを行っています(図3)。
二次元半導体点欠陥の第一原理計算
近年、ムーアの法則を乗り越え、次世代半導体素子のさらなる集積度の向上に向けて二次元半導体の研究が積極的に行われています。その中でも遷移金属ジカルコゲナイドは優れたキャリア移動度と絶縁性を保有していることから、二次元電子デバイスへの幅広い応用が期待されている物質です。
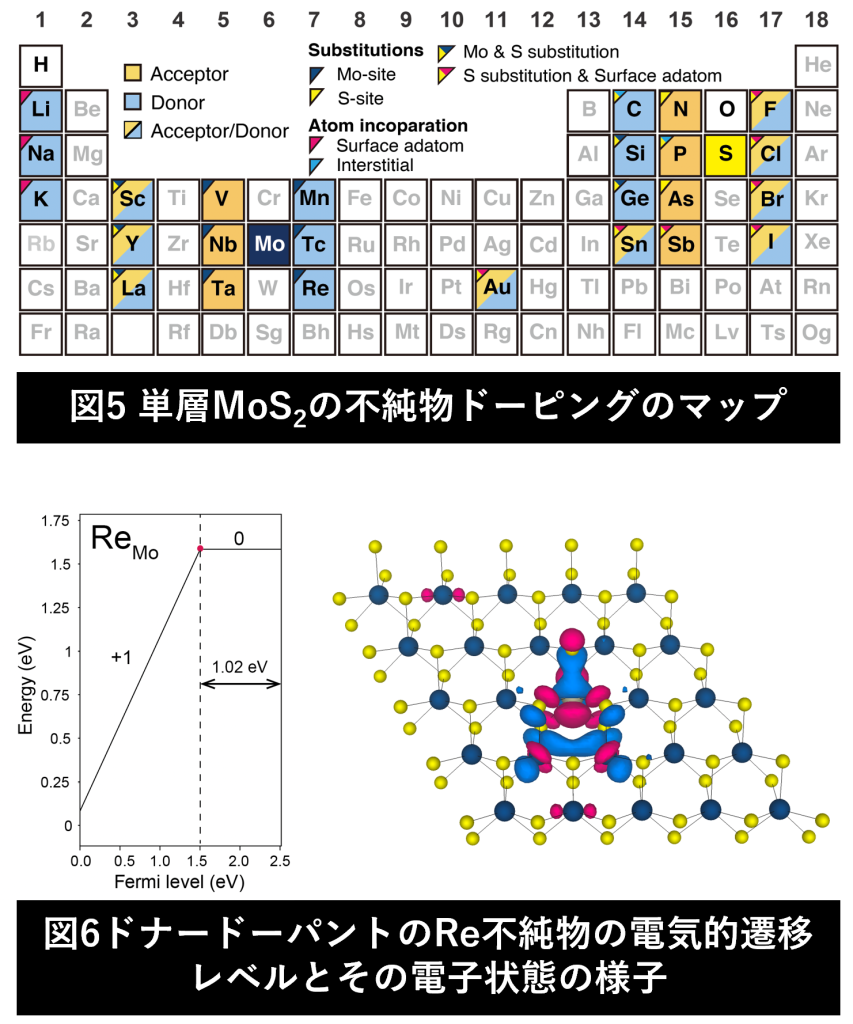
半導体では、不純物を添加する「ドーピング」により物性を制御可能です。「どのサイトにどのような元素がドーピングされるか」が半導体物性を決定づけるといっても過言ではありません(図5) 。第一原理計算を用いることで、ドーピングされた物質の電子状態を計算することが可能です(図6)。本研究室では遷移金属ジカルコゲナイドを中心とした二次元半導体の効果的な不純物ドーピングの方法を第一原理計算を用いて模索し、次世代の二次元デバイスの実現を目指しています。
第一原理分子動力学計算を用いた次世代電池材料探索
大きな放電容量を持ち、地殻中の豊富に存在するという理由から、Mgが次世代の二次電池材料として注目を集めています。Mg二次電池実現化には、高放電容量・高作動電圧・構造安定性・高イオン伝導度を持つ正極材料が必須となっています。しかし、Mgはイオン半径が大きい2価のイオンであるため、母結晶への強い親和性を持ち、これが構造安定性やイオン電導度に悪い影響を与えることが知られています。本研究室では、図7のようなハイスループットフローを用いてイオン伝導機構解明を試みています。さらに、不純物をドーピングすることで高い構造安定性・イオン電導度を持つ新規正極材を探索しています。
